The following is an example of global sequence alignment using Needleman/Wunsch techniques. For this example, the two sequences to be globally aligned are
G A A T T C A G T T A (sequence #1)
G G A T C G A (sequence #2)
So M = 11 and N = 7 (the length of sequence #1 and sequence #2, respectively)
An advanced scoring scheme is assumed where
The first step in the global alignment dynamic programming approach is to create a matrix with M + 1 columns and N + 1 rows where M and N correspond to the size of the sequences to be aligned.
The first row and first column of the matrix can be initially filled with 0.
One possible (inefficient) solution of the matrix fill step finds the maximum global alignment score by starting in the upper left hand corner in the matrix and finding the maximal score Mi,j for each position in the matrix. In order to find Mi,j for any i,j it is minimal to know the score for the matrix positions to the left, above and diagonal to i, j. In terms of matrix positions, it is necessary to know Mi-1,j, Mi,j-1 and Mi-1, j-1.
For each position, Mi,j is defined to be the maximum score at position i,j; i.e.
Mi,j = MAXIMUM[
Mi-1, j-1 + Si,j (match/mismatch in the diagonal),
Mi,j-1 + w (gap in sequence #1),
Mi-1,j + w (gap in sequence #2)]
Note that in the example, Mi-1,j-1 will be red, Mi,j-1 will be green and Mi-1,j will be blue.
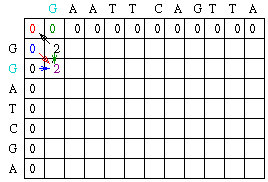
Using this information, the score at position 1,1 in the matrix can be calculated. Since the first residue in both sequences is a G, S1,1 = 2, and by the assumptions stated earlier, w = -2. Thus, M1,1 = MAX[M0,0 + 2, M1,0 - 2, M0,1 - 2] = MAX[2, -2, -2].
A value of 2 is then placed in position 1,1 of the scoring matrix. Note that there is also an arrow placed back into the cell that resulted in the maximum score, M[0,0].

Moving down the first column to row 2, we can see that there is once again a match in both sequences. Thus, S1,2 = 2. So M1,2 = MAX[M0,1 + 2, M1,1 - 2, M0,2 -2] = MAX[0 + 2, 2 - 2, 0 - 2] = MAX[2, 0, -2].
A value of 2 is then placed in position 1,2 of the scoring matrix and an arrow is placed to point back to M[0,1] which led to the maximum score.
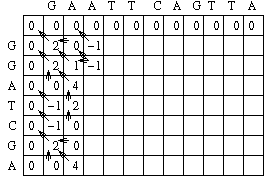
Looking at column 1 row 3, there is not a match in the sequences, so S 1,3 = -1. M1,3 = MAX[M0,2 - 1, M1,2 - 2, M0,3 - 2] = MAX[0 - 1, 2 - 2, 0 - 2] = MAX[-1, 0, -2].
A value of 0 is then placed in position 1,3 of the scoring matrix and an arrow is placed to point back to M[1,2] which led to the maximum score.

We can continue filling in the cells of the scoring matrix using the same reasoning.
Eventually, we get to column 3 row 2. Since there is not a match in the sequences at this positon, S3,2 = -1. M3,2 = MAX[ M2,1 - 1, M3,1 - 2, M2,2 - 2] = MAX[0 - 1, -1 - 2, 1 -2] = MAX[-1, -3, -1].

Note that in the above case, there are two different ways to get the maximum score. In such a case, pointers are placed back to all of the cells that can produce the maximum score.
The rest of the score matrix can then be filled in. The completed score matrix will be as follows:

The traceback step begins in the M,J position in the matrix, i.e. the position where both sequences are globally aligned.
Since we have kept pointers back to all possible predacessors, the traceback step is simple. At each cell, we look to see where we move next according to the pointers. To begin, the only possible predacessor is the diagonal match.

This gives us an alignment of
A
|
A
Note that the blue letters and gold arrows indicate the path leading to the maximum score.
We can continue to follow the path using a single pointer until we get to the following situation.
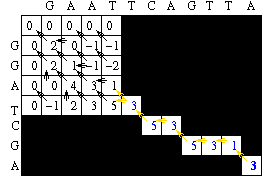
The alignment at this point is
T C A G T T A
| | | |
T C _ G _ _ A
Note that there are now two possible neighbors that could result in the current score. In such a case, one of the neighbors is arbitrarily chosen.
Once the traceback is completed, it can be seen that there are only two possible paths leading to a maximal global alignment.
One possible path is as follows:
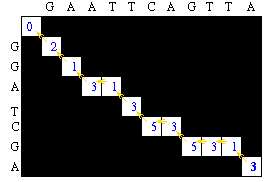
This gives an alignment of
G A A T T C A G T T A | | | | | | G G A _ T C _ G _ _ A
The other possible path is as follows:

This gives an alignment of
G A A T T C A G T T A | | | | | | G G A T _ C _ G _ _ A
Remembering that the scoring scheme is +2 for a match, -1 for a mismatch, and -2 for a gap, both sequences can be tested to make sure that they result in a score of 3.
G A A T T C A G T T A | | | | | | G G A _ T C _ G _ _ A + - + - + + - + - - + 2 1 2 2 2 2 2 2 2 2 2
2 - 1 + 2 - 2 + 2 + 2 - 2 + 2 - 2 - 2 + 2 = 3
G A A T T C A G T T A | | | | | | G G A T _ C _ G _ _ A + - + + - + - + - - + 2 1 2 2 2 2 2 2 2 2 2
2 - 1 + 2 + 2 - 2 + 2 - 2 + 2 - 2 - 2 + 2 = 3
so both of these alignments do indeed result in the maximal alignment score.